







Este estudio ofrece un análisis de las secuencias de genes por Repeticiones Palindrómicas Cortas Agrupadas Regularmente Interespaciadas (CRISPR, por sus siglas en inglés)/Cas en microorganismos de alta influencia en la producción acuícola asociados a infecciones en la especie Litopenaeus vannamei, cuyos resultados permiten proponer su uso en el tratamiento de infecciones causadas por la familia Vibrionaceae, debido a la baja ocurrencia de sistemas CRISPR en las especies estudiadas y la baja inmunidad a sus fagos, asegurando así una mayor sensibilidad que puede explorarse para el desarrollo de estrategias como la fagoterapia en el cultivo del camarón.
En el cultivo de camarón (L. vannamei), la familia de proteobacterias Vibrionaceae, el género Vibrio, representa uno de los principales responsables de las infecciones con alta influencia en la producción larvaria asociadas al cultivo de camarón. Las bacterias de este género se consideran parte de la microbiota normal del camarón, constituyendo el porcentaje más alto de todas las bacterias aisladas del tracto digestivo, branquias, cutículas y, ocasionalmente, en la hemolinfa.
Sin embargo, en condiciones de estrés o desequilibrio en la microbiota bacteriana natural, estas bacterias pueden inducir el desarrollo de infecciones en organismos como la vibriosis (o septicemia bacteriana del camarón).
Las principales cepas virulentas de camarón, del género Vibrio, identificadas en varios estadios larvales son V. alginolyticus, V. anguillarum, V. parahaemolyticus, V. harveyi y V. vulnificus, las cuales generan altas mortalidades y pérdidas económicas en la producción.
La prevención y control de enfermedades en la camaronicultura se basa principalmente en el uso de antibióticos; sin embargo, la creciente aparición de cepas bacterianas resistentes a estos, sumado a la preocupación inducida por la perspectiva de no contar con antibióticos efectivos en un futuro próximo, ha impulsado la búsqueda de nuevas alternativas antagónicas hacia aquellas que aún no han desarrollado mecanismos de resistencia, con menores efectos secundarios, y con alta especificidad, para evitar la alteración de la microbiota intestinal del ser humano y garantizar una mayor efectividad de los tratamientos.
“Aunque no está claro si existe algún mecanismo de resistencia bacteriana frente a enzibióticos (fago lisinas), “Repeticiones palindrómicas cortas agrupadas regularmente interespaciadas” (CRISPR, por sus siglas en inglés) han mostrado una distintiva característica en la mayoría de los genomas de bacterias y arqueas, que confieren resistencia contra bacteriófagos.”
Una de las estrategias prometedoras en el camino hacia el objetivo de encontrar antagonistas más eficaces, menos tóxicos y específicos para el control de los microorganismos causantes de infecciones, está representada por los virus que infectan exclusivamente a las bacterias, llamados bacteriófagos, y sus derivados enzimáticos (enzibióticos). Se trata de agentes inhibidores que se han descrito como potenciales agentes antimicrobianos con interés terapéutico, incluida su aplicación en el cultivo de camarón.
Aunque no está claro si existe algún mecanismo de resistencia bacteriana frente a enzibióticos (fago lisinas), CRISPR han mostrado una distintiva característica en la mayoría de los genomas de bacterias y arqueas, que confieren resistencia contra bacteriófagos.
El objetivo de este estudio, consistió en realizar un análisis comparativo de Sistemas CRISPR en genomas de microorganismos con influencia patógena en camarón Litopenaeus vannamei.
Secuencias genómicas e identificación de estructuras CRISPR
Se estudiaron cinco especies de Vibrio: Vibrio alginolyticus, V. anguillarum, V. parahaemolyticus, V. harveyi, V. vulnificus y la especie Photobacterium damselae, todas asociadas a infecciones sistémicas en camarones, como la vibriosis.
Al respecto, 69 de las secuencias corresponden a secuencias genómicas completas, las cuales incluyen 14 de V. alginolyticus, 13 de V. anguillarum, 21 de V. parahaemolyticus, 4 de V. harveyi, 15 de V. vulnificus, así como 2 secuencias cromosómicas completas de Photobacterium damselae.
Las secuencias genómicas públicas de los loci CRISPR se obtuvieron de la base de datos CRISPRdb.
“El 53% de los espaciadores (75/142) presentaron homología con material genético extracromosómico, mientras que el 47% restante de las secuencias espaciadoras (67/142) exhibieron homología con bacteriófagos.”
Alternativamente, los loci CRISPR en los genomas se identificaron a través de CRISPRFinder. A tal efecto, se siguieron dos criterios de búsqueda. Uno de ellos fue el cribado de posibles ubicaciones de CRISPR mediante la detección de repeticiones máximas (repetición con la máxima extensión posible a la derecha o izquierda sin incurrir en un desajuste) con el uso del paquete VMatch, que es la actualización de REPuter basada en una implementación eficiente de arreglos de sufijos mejorados.
Los parámetros predeterminados utilizados fueron los siguientes: una longitud de repetición de 23 a 55 pb, un tamaño de espacio entre repeticiones de 25 a 60 pb, un 20% de desajuste de nucleótidos entre repeticiones.
“El 92% de las secuencias espaciadoras únicas encontradas en los loci CRISPR presentaron homología con alguna secuencia contenida en la base de datos GenBank contra material genético extracromosómico (plásmidos) o bacteriófagos, lo que es indicativo de una clara función de inmunidad contra material genético extraño y de una alta especificidad de los diferentes sistemas CRISPR/Cas estudiados.”
El otro criterio de búsqueda se basó en la determinación de la función CRISPR, para lo cual se agregan filtros con la finalidad de ayudar a validar un CRISPR, como la búsqueda de espaciadores no idénticos con un tamaño que debe ser de 0.6 * a 2.5 * el tamaño de la repetición. Este filtro está configurado para eliminar repeticiones en tándem.
La comparación de los espaciadores se realizó alineándolos (utilizando los parámetros predeterminados del programa Muscle). El porcentaje de similitud de los espaciadores se calcula con la función percent_identity de la interfaz (Bio) perl (métodos AlignIO, interfaz Muscle) y un parámetro establecido en 60%.
Para discriminar entre estructuras CRISPR confirmadas de aquellas cuestionables, las estructuras pequeñas similares a CRISPR, se clasifican utilizando un nivel de evidencia, calificado de 1 a 4, donde 1 incluye pequeños CRISPR (con tres o menos espaciadores) y 2 a 4 se catalogan en función de la repetición y similitud del espaciador.
Análisis bioinformático y resultados estadísticos
Para la búsqueda de genes Cas, el primer paso consistió en la identificación de marcos de lectura abiertos (ORF). Los árboles filogenéticos se generaron con base en el método de grupos de pares no ponderados (UPGMA) para la proteína central Cas, cuya estructura se evaluó mediante múltiples alineamientos de secuencia con alineamiento global Geneious.
Para establecer diferencias entre los parámetros comparativos considerados se utilizó el análisis de varianza de un factor (ANOVA) como modelo estadístico.
Organización y diversidad de sistemas CRISPR / Cas en el genoma de Vibrionaceae
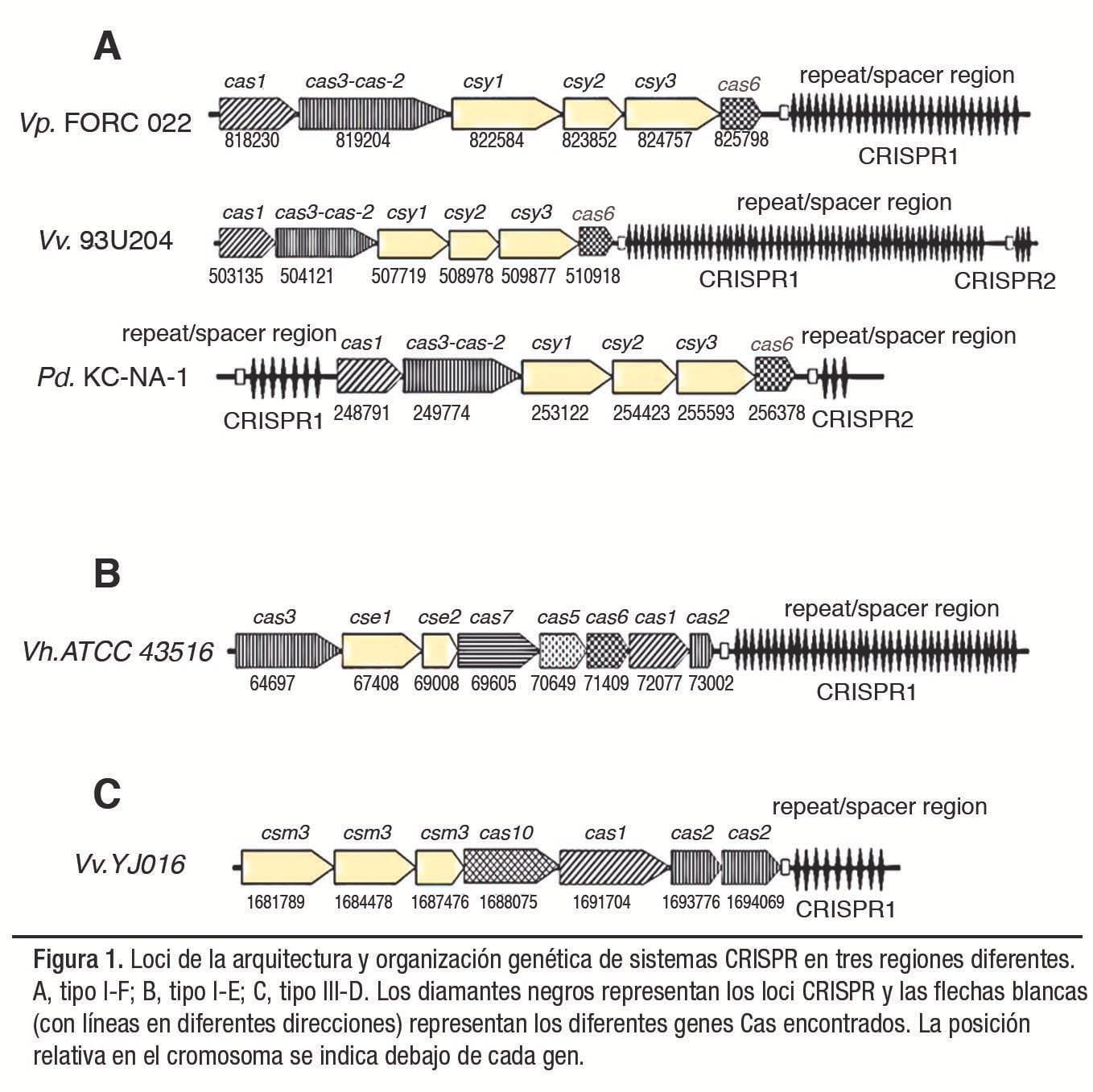
Se encontraron siete estructuras CRISPR en cinco genomas dentro del total (1,104 genomas) de la familia de proteobacterias Vibrionaceae estudiadas, distribuidas en tres especies de Vibrio (V. parahaemolyticus FORC_022, V. harveyi ATCC 43516, V. vulnificus YJ016 y 93U204) y en la especie relacionada Photobacterium damselae KC-Na-1. Así mismo, se detectaron tres matrices CRISPR/ Cas diferentes en Vibrio que tienen diferentes ubicaciones en los cromosomas (Figura 1).
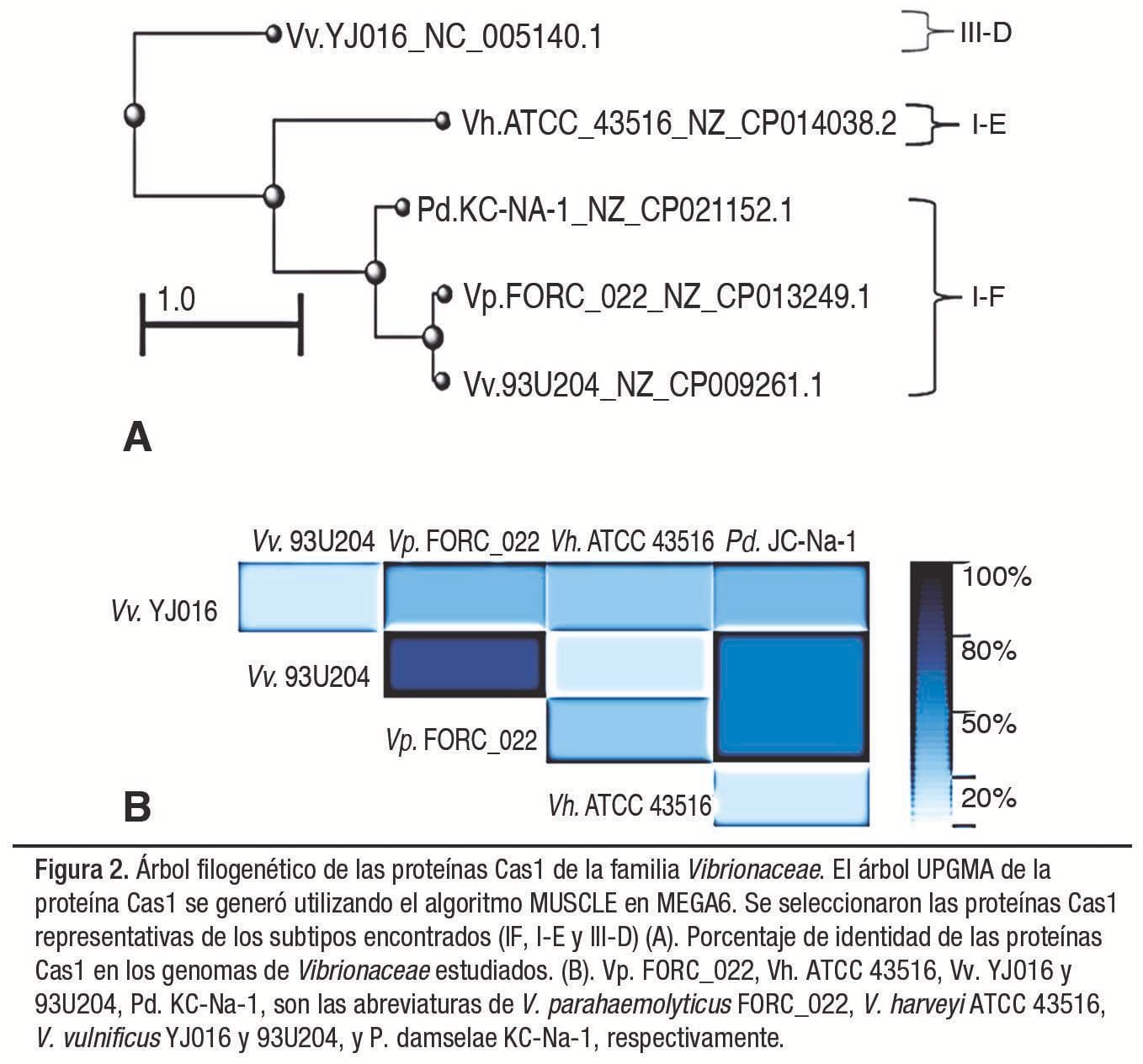
Para distinguir mejor el sistema CRISPR/Cas en el género Vibrio, o en los miembros con matrices CRISPR encontradas en la familia Vibrionaceae, se construyó un árbol filogenético basado en la homología de las proteínas Cas1 de cada especie (Figura 2). También, se evaluó la distinción entre el tipo de CRISPR encontrado por el análisis de secuencia de repetición directa (DR). Se encontró que las DR estaban interespaciadas, con un mínimo de 3 a un máximo de 55 secuencias espaciadoras de una longitud similar entre 31 y 43 pb.
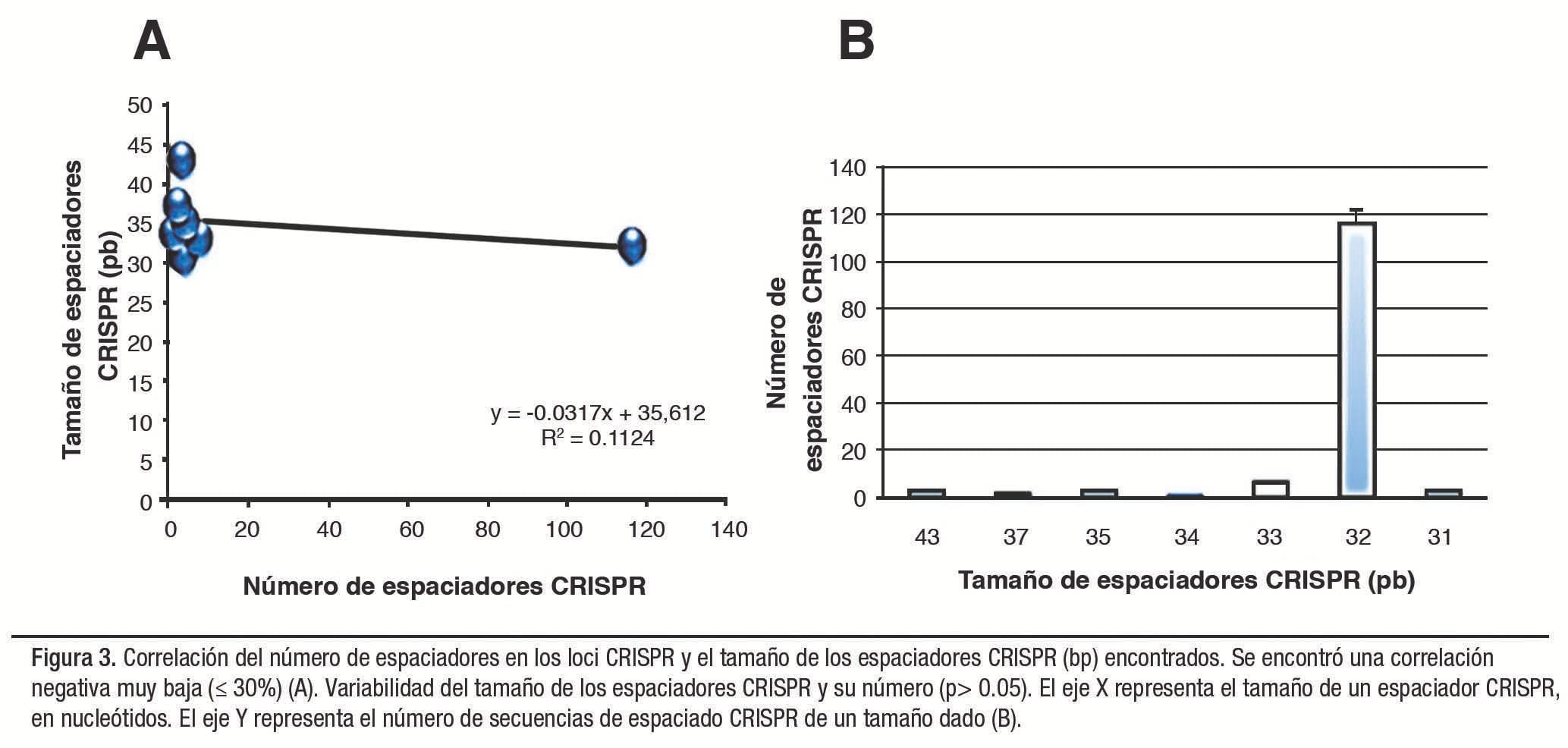
A pesar de que la mayoría de las secuencias espaciadoras presentaron un tamaño de 32 pb, el análisis de varianza indica que no existe una diferencia estadísticamente significativa (p>0.01) que permita señalar que las medias obtenidas correspondientes al número y tamaño de las secuencias espaciadoras son diferentes entre sí.
Sin embargo, existe una correlación muy baja entre el número y el tamaño que presentan estas secuencias (Figura 3). El número total de espaciadores detectados por tipo de CRISPR fue 96/142 para los tipos I-F (68%), 37/142 para el tipo I-E (26%) y 9/142 para el tipo III-D (6%). Las estructuras CRISPR observadas exhibieron entre 6 y 8 genes asociados con las secuencias CRISPR (Cas).
La presencia de subtipos I-F corresponde a lo descrito en la literatura en cepas del género como V. cholerae O1/O139, en las que, tras análisis bioinformático, se han estudiado módulos CRISPR/Cas los cuales contienen los genes que codifican las proteínas Cas1, Cas3 identificados y Cas6, y en base a las secuencias de proteínas y su organización, se han agrupado en sistemas CRISPR/Cas de tipo IF.
En la especie V. harveyi ATCC 43516 únicamente se detectó una estructura CRISPR confirmada, como la descrita en cepas como V. cholerae O395. Curiosamente, se encontró una nueva forma de disposición de genes Cas para el subtipo III-D detectado en V. vulnificus YJ016.
El origen de los espaciadores CRISPR
El 53% de los espaciadores (75/142) presentaron homología con material genético extracromosómico, mientras que el 47% restante de las secuencias espaciadoras (67/142) exhibieron homología con bacteriófagos. Estos resultados indican una clara función de inmunidad frente a material genético extraño (plásmidos o fagos) y una alta especificidad de los diferentes sistemas CRISPR/Cas estudiados.
Las homologías evidenciadas por las secuencias espaciadoras en estudio indicaron una alta diversidad en términos de las dianas de reconocimiento de los sistemas de inmunidad CRISPR examinados, representada por la homología con secuencias de DNA relacionadas o no con representantes de la familia Vibrionaceae.
El 92% de las secuencias espaciadoras únicas encontradas en los loci CRISPR presentaron homología con alguna secuencia contenida en la base de datos GenBank contra material genético extracromosómico (plásmidos) o bacteriófagos, lo que es indicativo de una clara función de inmunidad contra material genético extraño y de una alta especificidad de los diferentes sistemas CRISPR/Cas estudiados.
Conclusión
Los resultados obtenidos en esta investigación son muy prometedores porque muestran una baja ocurrencia de estructuras CRISPR en estas especies Vibrionaceae con inmunidad a fagos líticos específicos del género Vibrio, lo cual es importante si se considera que esta sensibilidad puede ser explorada para el desarrollo de estrategias como la fagoterapia en el cultivo de L. vannamei, una tecnología basada en el uso de enzimas o fagos para el control de patógenos típicos del camarón, reduciendo de esta manera las pérdidas ocasionadas por infecciones de las granjas camaroneras.
Esta es una versión resumida desarrollada por el equipo editorial de Panorama Acuícola Magazine del artículo “CHARACTERIZATION OF CRISPR GENETIC SEQUENCES IN MICROORGANISMS ASSOCIATED WITH INFECTIONS IN SHRIMP (LITOPENAEUS VANNAMEI)” escrito por ÁNGEL PARRA, CARLA LOSSADA, ALEIVI PÉREZ, JOHNNY NAVARRETE, AND LENIN GONZÁLEZ.
La versión original fue publicada en JUNIO de 2020 a través REVISTA DE LA FACULTAD DE AGRONOMÍA DE LA UNIVERSIDAD DEL ZULIA. Se puede acceder a la versión completa a través de https://doi.org/10.47280/ RevFacAgron(LUZ).v38.n2.08





